 手机版
手机版
 关于我们
关于我们 加入收藏
加入收藏

步琦实验室设备贸易(上海)有限公司
5 年 白金会员
白金会员
白金会员 已认证
已认证
400-810-0069转3366
获取底价
提交后,商家将派代表为您专人服务
步琦实验室设备贸易(上海)有限公司
白金会员已认证

利用步琦SFC系统纯化
阿司匹林、苯佐卡因和普鲁卡因
SFC应用
1
简介
化学合成常伴有杂质,因产率很少达 100%,杂质会严重影响药物疗效、安全和质量,所以纯化药物保障其纯度和完整性至关重要,在现代药物研发的过程中往往通过色谱法等多种方法纯化。
在上一篇应用文章《利用步琦 SFC 系统纯化利多卡因与乙酰氨基酚》中,我们使用超临界流体色谱(SFC)高效纯化了利多卡因和乙酰氨基酚的合成产物。在此过程中,我们先使用步琦独有的 Sepmatix 8x SFC 平行色谱系统快速筛选了色谱柱,之后将其放大到 BUCHI Sepiatec SFC-50 超临界制备色谱系统上。本次应用中,我们依旧会使用同样的工作流程去纯化阿司匹林、苯佐卡因和普鲁卡因的合成产物。
2
实验设备
BUCHI Sepmatix 8x SFC 8通道平行色谱系统
BUCHI Sepiatec SFC-50 超临界制备色谱系统
BUCHI PrepPure 硅胶,5um,250×4.6mm
BUCHI PrepPure 二醇基,5um,250×4.6mm
BUCHI PrepPure 氨基,5um,250×4.6mm
BUCHI PrepPure 2-EP,5um,250×4.6mm
HILIC柱,5um,250×4.6mm (Dr. Maisch GmbH)
BUCHI PrepPure PEI,5um,250×4.6mm
BUCHI PrepPure PEI,5um,250×4.6mm
BUCHI PrepPure CBD,5um,250×4.6mm
氰基柱,5um,250×10mm ,(Dr. Maisch GmbH)
BUCHI PrepPure 2-EP,5um,250×10mm
BUCHI PrepPure 二醇基,5um,250×10mm
3
化学品与样品
化学品:
二氧化碳 (99.9%)
甲醇 (≥99%)
甲醇溶液中 2M 的氨溶液
甲酸(99%)
去离子水
为了安全处理,请注意所有相应的MSDS!
样品:
普鲁卡因合成产物
阿司匹林合成产物
苯佐卡因合成产物
4
程序设定
BUCHI Sepmatix 8x SFC 平行色谱柱系统
流动相:A= 二氧化碳;B= 甲醇
柱尺寸:250×4.6mm
流速:3mL/min(每根色谱柱)
检测:DAD 紫外扫描 200 nm - 600 nm
流动相条件:
0 – 0.5 min | 5 % B | |
0.5 – 8.0 min | 5 – 50 % B | |
8.0 – 9.4 min | 50 % B | |
9.4 – 9.5 min | 50 – 5 % B | |
9.5 – 10 min | 5 % B | |
筛选运行完全自动运行,流速设置为 3mL/min 每通道,使用流控单元,平衡每一根色谱柱。样品自动注入(V=5μL),并开始平行筛选(运行时间=10min)。背压调节器设置为 150 bar,柱子加热至 32℃,可按需往改性剂中加入添加剂改善峰型。
BUCHI Sepiatec SFC-50 超临界制备色谱系统
流动相:A= 二氧化碳;B= 甲醇
柱尺寸:250×10mm
流动相条件:等度运行条件
检测:紫外
所有 10mm ID 色谱柱都在预设流速下平衡 3 分钟,使用自动进样器上样,并开始运行。背压调节器设置为 150 bar,柱子加热至 40℃,可按需往改性剂中加入添加剂改善峰型。
5
结果
5.1 阿司匹林
阿司匹林,化学名为乙酰水杨酸(下称 ASA),是一种众所周知的镇痛和抗炎药物。它属于非甾体抗炎药(NSAIDs)这一类。可以通过将水杨酸(下称 SA)与乙酸酐发生酯化反应而化学合成(见图5) [1] 。为了确定乙酰水杨酸的理想分离选择性,首先进行了色谱柱柱筛选(见图1)。
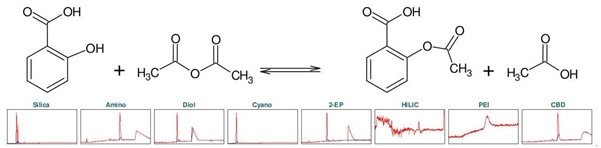
▲ 图 1:顶部:阿司匹林合成的反应方程式,底部:Sepmatix 8x SFC 仪器色谱柱筛选结果;从左到右:硅胶,氨基,二醇基,氰基,2-EP,HILIC,PEI和CBD;运行时间 = 10分钟。
由于 SA 在固定相中的保留性较高,为了强制洗脱 SA,在梯度中增加了一个等度步骤,即在 50% 改性剂浓度下进行 5 分钟的等度洗脱。在 HILIC 相中,ASA 和 SA 只有噪音而无洗脱。在 PEI 相中,在实验条件下只有 ASA 被洗脱。除氰基相外,其余各相均显示出 ASA 和 SA 的分离。结合分离时长和分辨率而言,2-EP 相的结果最好(见表 1)
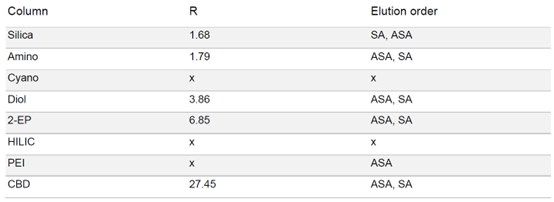
▲ 表1:样品在不同固定相色谱柱条件下的分辨率值和洗脱顺序
选择 2-EP 相作为固定相,放大至制备规模。在色谱柱筛选的过程中,ASA 和 SA 在 2-EP上被洗脱的甲醇比例为 26 - 40%。图 2(上)显示了在甲醇比例为 40% 的10x250mm 2-EP 色谱柱上纯化 ASA 和 SA 的情况。改性剂中添加了甲酸(0.5%),以改善 SA 的峰形。在不添加甲酸的情况下,SA 的拖尾非常严重。在相同条件下,可采用叠加进样法自动纯化 ASA(见图2下),并进行馏分收集。
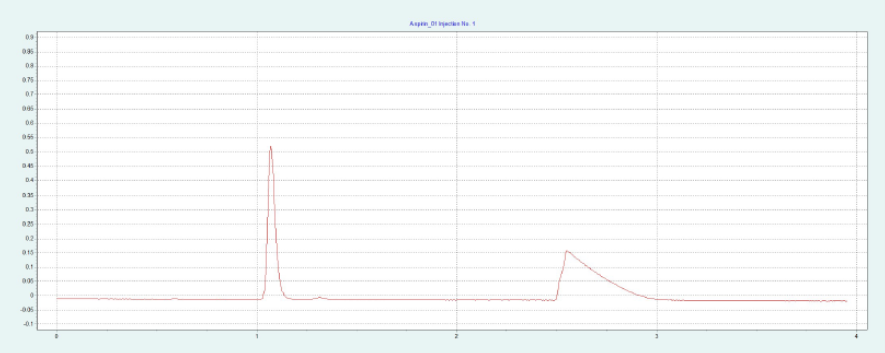
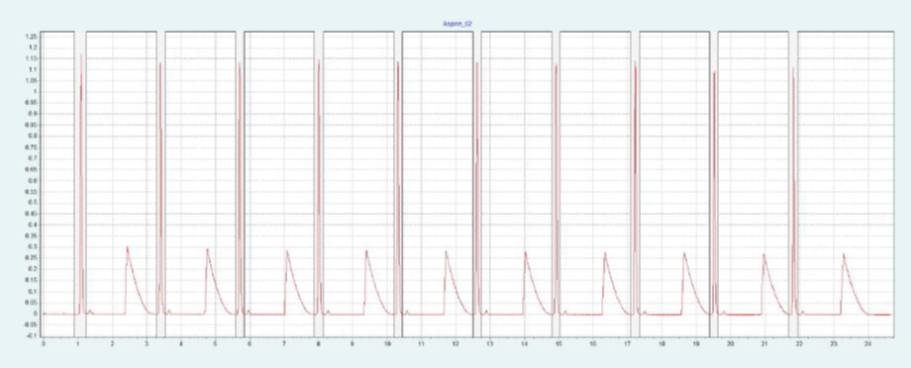
▲ 图 2:ASA 纯化的单次进样(上图)和叠加进样(下图);运行条件:流速 = 30 mL/min,改性剂 = 甲醇 + 0.5 % 甲酸,改性剂 % = 40 %,温度 = 40 °C,压力 BPR = 150 bar,进样量 = 200 μL,紫外波长 = 254 nm;叠加进样条件:进样次数 = 10,叠加时间 = 2.0 min,馏分 = 1(基于时间)。
5.2 苯佐卡因
苯佐卡因(下称 BC),化学名为对氨基苯甲酸乙酯,属于局部麻醉剂类。它可以从对氨基苯甲酸(下称 PABA)通过酸催化羧基与乙醇的酯化反应化学合成,用硫酸作为催化剂 [2]。首先进行色谱柱筛选,以确定苯佐卡因合成产物理想的分离选择性(见 图3)。
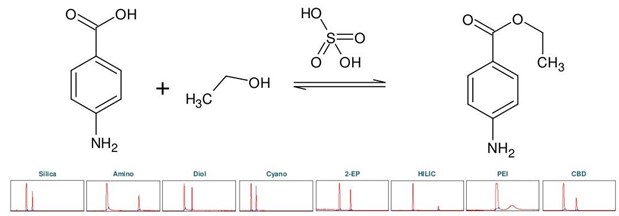
▲ 图3:顶部:苯佐卡因合成反应方程式,底部:Sepmatix 8x SFC仪器色谱柱筛选结果;从左到右:硅胶,氨基,二醇基,氰基,2-EP,HILIC,PEI和CBD;运行时间 = 15分钟。
由于 PABA 在固定相中的保留性较强,因此在梯度中增加了一个等度步骤,在 50% 甲醇的条件下运行 5 分钟,硫酸通过洗涤步骤去除避免过高的酸性。各固定相的色谱图显示,每种固定相都能分离苯佐卡因,但是洗脱速度和分辨率存在很大差异(见表2)。HILIC 和氨基相的分辨率最高,但 PABA 的保留时间过长,即使用50%的甲醇洗脱,依旧需要很长时间。
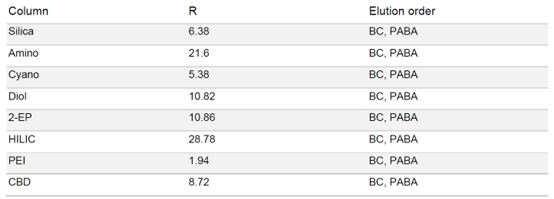
▲ 表2:样品在不同固定相色谱柱条件下的分辨率值和洗脱顺序。
结合柱筛选结果,最终选择二醇基相作为制备色谱柱。HILIC 和氨基相需要较高比例的甲醇和较长的运行时间。二醇基相不仅可以非常快速地洗脱样品,并具有足够高的分辨率。二醇基柱的筛选结果表明,BC 和 PABA 在甲醇比例为 30 - 40% 时即可洗脱。
图 4(上图)显示了 10 x 250mm 的二醇基色谱柱在甲醇比例为 28% 时对苯佐卡因的纯化情况。在相同条件下,可采用叠加进样法自动纯化 BC(见图4下),并进行馏分收集。
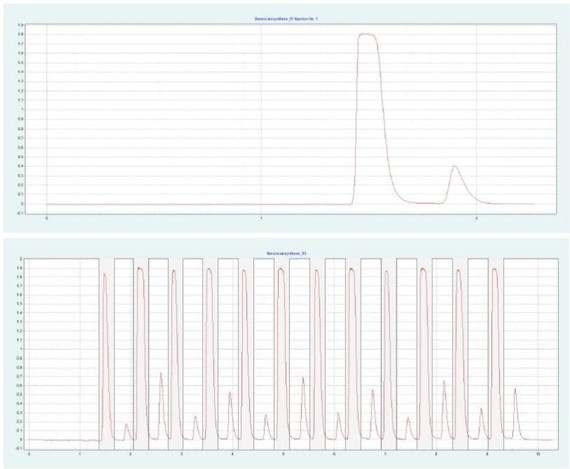
▲ 图 4:苯佐卡因的单次注射(顶部)和堆叠注射(底部)纯化;运行条件:流速 = 20 mL/min, 改性剂 = 甲醇,改性剂 % = 28%,温度 = 40 °C,压力 BPR = 150 bar,注射量 = 90 μL,UV 波长 =276nm;堆叠注射条件:注射次数 =12,堆叠时间 =0.66min,馏分 =1(基于时间的)。
5.3 普鲁卡因
普鲁卡因(下称 PC),化学名为 4-氨基苯甲酸 2-(N, N-二乙基氨基)乙酯,是一种药物,具有局部麻醉作用。它阻断电压依赖性钠离子通道,从而导致例如疼痛敏感性的降低。普鲁卡因可以通过化学方法在碱性条件下从 4-氨基苯甲酸乙酯(下称ABE)合成。这涉及到使用 2-二乙基氨基乙醇进行酯交换[3]。碱是乙醇钠醇,它可以通过钠与乙醇的反应产生。为确定普鲁卡因合成产物纯化的理想选择性,进行了色谱柱筛选(见图 5)。
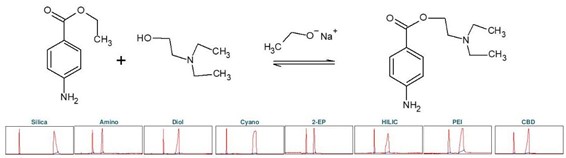
▲ 图5:顶部:普鲁卡因合成的反应方程式,底部:Sepmatix 8x SFC 仪器色谱柱筛选结果;从左到右:硅胶,氨基,二醇基,氰基,2-EP,HILIC,PEI 和 CBD;运行时间 = 15分钟。
由于普鲁卡因在固定相中的保留性较高,因此在梯度中增加了一个等度步骤,即在 50% 甲醇浓度下进行 5 分钟的梯度。结果显示,每种固定相对普鲁卡因的纯化都有合适的选择性(见表3)。普鲁卡因在硅胶、PEI 和氰基固定相中的保留性较高。
由于普鲁卡因的基本特性,在部分色谱柱上的峰形非常不对称。二醇基相下的峰前沿非常明显,不对称度系数为 0.58,而硅胶相下的拖尾非常明显,不对称度系数为 2.73。
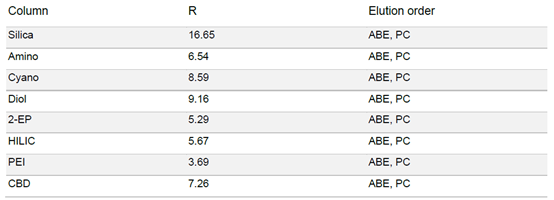
▲ 表3:样品在不同固定相色谱柱条件下的分辨率值和洗脱顺序。
结合柱筛选结果,选择 2-EP 相放大为制备纯化的方法。硅胶相需要高含量的甲醇才能洗脱出 PC,导致运行时间长,峰形不佳。而 2-EP 相既可以快速洗脱样品,又具有足够高的分辨率。为了改善峰型,在甲醇改性剂中加入 5 % 的去离子水和 20 mM 的氨水作为添加剂。
筛选结果表明,PC 和 ABE 在改性剂含量约为 24 - 32% 时可以从色谱柱内洗脱下来。图6(上图)显示了10 x 250mm 的 2-EP 色谱柱在改性剂含量为 25% 时纯化 PC 的情况。在相同条件下,可采用叠加进样法自动纯化 PC(见图6,下图)并收集馏分,添加添加剂可显著改善 PC 的峰形。
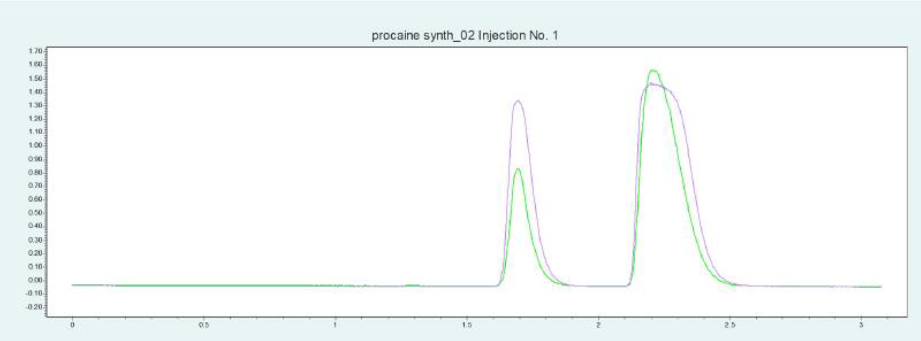
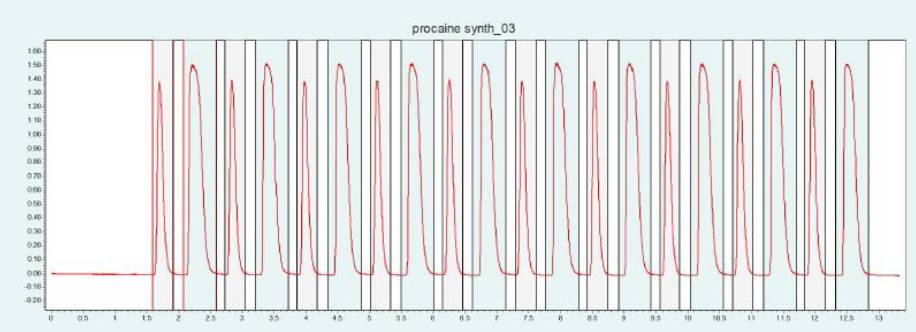
▲ 图 6:普鲁卡因纯化的单次进样(上图)和叠加进样(下图);运行条件:流速 = 20 mL/min,改性剂 = 甲醇/水(95/5 %)加 20 mM 氨水,改性剂 % = 25 %,温度 = 40 °C,压力 BPR = 150 bar,进样量 = 100 μL,紫外波长 = 220 nm;叠加进样条件:进样次数 = 10,叠加时间 = 1.1min,馏分 = 2(基于时间)。
6
结论
超临界制备色谱纯化合成产物高效快速,但需筛选合适的色谱填料。BUCHI Sepmatix 8x SFC 平行色谱系统可快速筛选填料并放大至制备规格,既能提高样品分离度,又能充分利用色谱柱上样量,为 BUCHI Sepiatec SFC-50 制备系统的叠加进样奠定良好基础,二者结合可达到降本增效的目的。
7
参考文献
Th. Eicher und H. J. Roth; Synthese, Gewinnung und Charakterisierung von Arzneistoffen, Georg Thieme Verlag, Stuttgart (1986).
Winterfeld, K. – Praktikum der organisch-präparativen Pharmazeutischen Chemie, 6. Auflage, Steinkopff Verl., Darmstadt (1965).
Axel Kleemann, Jürgen Engel, Bernd Kutscher und Dietmar Reichert: Pharmaceutical Substances, 4. Auflage, Georg Thieme Verlag, Stuttgart (2000).

长按上方二维码联系我们
或拨打联系电话:
400 - 880 - 8720
微信公众号
步琦智慧实验室
淘宝官方旗舰店
瑞士步琦
最新动态
更多





请拨打厂商400电话进行咨询
使用微信扫码拨号


 上一篇
上一篇